Create genetic perturabation data
1. import the necessary libraries
[1]:
import xpert as xp
from xpert.data.utils import get_info_txt
import scanpy as sc
import os
import pickle
import warnings
import logging
warnings.filterwarnings('ignore')
sc.settings.verbosity = 0
logging.getLogger('scanpy').setLevel(logging.ERROR)
logging.getLogger('anndata').setLevel(logging.ERROR)
2. load original adata file of perturbation data
[2]:
adata = sc.read('../../data/Norman2019/NormanWeissman2019_filtered.h5ad')
adata
[2]:
AnnData object with n_obs × n_vars = 111445 × 33694
obs: 'guide_id', 'read_count', 'UMI_count', 'coverage', 'gemgroup', 'good_coverage', 'number_of_cells', 'tissue_type', 'cell_line', 'cancer', 'disease', 'perturbation_type', 'celltype', 'organism', 'perturbation', 'nperts', 'ngenes', 'ncounts', 'percent_mito', 'percent_ribo'
var: 'ensemble_id', 'ncounts', 'ncells'
add obs.column with ‘perturbation_new’ and ‘cell_type_new’, which is used to perform data proprecessing.
[3]:
def get_perturbation(x):
return '; '.join(x['perturbation'].split('_'))
adata.obs['perturbation_new'] = adata.obs.apply(get_perturbation, axis=1)
adata.obs['celltype_new'] = adata.obs['celltype']
def get_perturbation_group(x):
return ' | '.join([x['perturbation_new'], x['celltype_new']])
perturbation_group = adata.obs.apply(get_perturbation_group, axis=1)
adata.obs['perturbation_group'] = perturbation_group
adata.obs['sgRNA_new'] = 'control'
3. generate Pert_Data object
[4]:
# parameters
pert_cell_filter = 100 # this is used to filter perts, cell number less than this will be filtered
seed = 2024 # this is the random seed
split_type = 1 # 1 for unseen perts; 0 for unseen celltypes
split_ratio = [0.7, 0.2, 0.1] # train:test:val; val is used to choose data, test is for final validation
var_num = 5000 # selecting hvg number
num_de_genes = 20 # number of de genes
bs_train = 32 # batch size of trainloader
bs_test = 32 # batch size of testloader
data_dir = '../../data/Norman2019/'
[5]:
# Create Pert_Data object
pert_data = xp.data.Byte_Pert_Data(
prefix='Norman2019',
pert_cell_filter=pert_cell_filter,
seed=seed,
split_ratio=split_ratio,
split_type=split_type,
var_num=var_num,
num_de_genes=num_de_genes,
bs_train=bs_train,
bs_test=bs_test
)
# Complete data processing pipeline
print("Step 1: Reading files...")
pert_data.read_files(adata)
print("Step 2: Filtering perturbations...")
pert_data.filter_perturbation()
print("Step 3: Preprocessing adata and selecting HVGs...")
pert_data.get_and_process_adata(var_num=pert_data.var_num)
print("Step 4: Setting control barcodes...")
pert_data.set_control_barcode()
print("Step 5: Calculating E-distances...")
pert_data.get_edis_2()
print("Step 6: Filtering sgRNAs...")
pert_data.adata_split.obs['sgRNA_new'] = 'control'
pert_data.filter_sgRNA()
print("Step 7: Data splitting...")
pert_data.data_split(split_type=1, test_perts=None)
print("Step 8: Getting differential genes...")
pert_data.get_de_genes()
print("Step 9: Saving processed data...")
# Save the processed data
pickle.dump(pert_data, open(os.path.join(data_dir, 'pert_data.pkl'), 'wb'))
print("Pert_Data object generation completed!")
print(f"Final dataset shape: {pert_data.adata_split.shape}")
print(f"Number of perturbation groups: {len(pert_data.filter_perturbation_list)}")
Step 1: Reading files...
========== read file finished!
Step 2: Filtering perturbations...
retain_pert_num is: 225
filtered pert num is: 12
100%|██████████| 236/236 [00:03<00:00, 73.45it/s]
========== filter perturbation finished!
Step 3: Preprocessing adata and selecting HVGs...
len of exclude_var_list is 4
len of pert_gene_list is 343
len of final var_names is 5040
========== get var genes finished!
this is new version
Step 4: Setting control barcodes...
100%|██████████| 224/224 [00:03<00:00, 69.05it/s]
========== set control barcodes finished!
Step 5: Calculating E-distances...
Step 6: Filtering sgRNAs...
0it [00:00, ?it/s]
Step 7: Data splitting...
========== data split finished!
Step 8: Getting differential genes...
100%|██████████| 224/224 [01:04<00:00, 3.48it/s]
========== get de genes finished!
Step 9: Saving processed data...
Pert_Data object generation completed!
Final dataset shape: (110554, 5040)
Number of perturbation groups: 224
4. Pert_Data Information Analysis and Visualization
[ ]:
# =============================================================================
# Pert_Data Information Analysis and Visualization
# =============================================================================
import matplotlib.pyplot as plt
import seaborn as sns
import numpy as np
import pandas as pd
# Set plotting style
plt.style.use('default')
sns.set_palette("husl")
# --------- 1. 分布相关的两个统计图分开画 ---------
print("1. Analyzing cell count distribution...")
pert_cell_counts = []
pert_names = []
for pert in pert_data.filter_perturbation_list:
cell_count = len(pert_data.adata_split[pert_data.adata_split.obs['perturbation_group'] == pert])
pert_cell_counts.append(cell_count)
pert_names.append(pert.split(' | ')[0]) # 只取gene name
mean_cells = np.mean(pert_cell_counts)
median_cells = np.median(pert_cell_counts)
# 单独画:每个扰动的细胞数量分布(直方图)
fig1, ax1 = plt.subplots(figsize=(7, 5))
ax1.hist(pert_cell_counts, bins=30, alpha=0.7, edgecolor='black')
ax1.set_xlabel('Number of Cells per Perturbation')
ax1.set_ylabel('Frequency')
ax1.set_title('Distribution of Cell Counts per Perturbation')
ax1.grid(True, alpha=0.3)
ax1.axvline(mean_cells, color='red', linestyle='--', label=f'Mean: {mean_cells:.1f}')
ax1.axvline(median_cells, color='orange', linestyle='--', label=f'Median: {median_cells:.1f}')
ax1.legend()
plt.tight_layout()
plt.show()
print(f" - Mean cells per perturbation: {mean_cells:.1f}")
print(f" - Median cells per perturbation: {median_cells:.1f}")
print(f" - Total perturbations: {len(pert_cell_counts)}")
# --------- 2. E-distance分布(单独画) ---------
print("\n2. Analyzing E-distance distribution...")
fig2, ax2 = plt.subplots(figsize=(7, 5))
if hasattr(pert_data, 'filter_pert_edis') and pert_data.filter_pert_edis:
edis_values = list(pert_data.filter_pert_edis.values())
ax2.hist(edis_values, bins=30, alpha=0.7, edgecolor='black', color='green')
ax2.set_xlabel('E-distance')
ax2.set_ylabel('Frequency')
ax2.set_title('Distribution of E-distances')
ax2.grid(True, alpha=0.3)
mean_edis = np.mean(edis_values)
median_edis = np.median(edis_values)
ax2.axvline(mean_edis, color='red', linestyle='--', label=f'Mean: {mean_edis:.3f}')
ax2.axvline(median_edis, color='orange', linestyle='--', label=f'Median: {median_edis:.3f}')
ax2.legend()
print(f" - Mean E-distance: {mean_edis:.3f}")
print(f" - Median E-distance: {median_edis:.3f}")
print(f" - Min E-distance: {np.min(edis_values):.3f}")
print(f" - Max E-distance: {np.max(edis_values):.3f}")
else:
ax2.text(0.5, 0.5, 'E-distance data not available',
ha='center', va='center', transform=ax2.transAxes)
ax2.set_title('E-distance Distribution (Not Available)')
print(" - E-distance data not available")
plt.tight_layout()
plt.show()
# --------- 3. UMAP单独画 ---------
print("\n3. Generating UMAP plot for specific perturbation...")
if pert_cell_counts:
# Get perturbations sorted by cell count
pert_cell_df = pd.DataFrame({
'perturbation': pert_data.filter_perturbation_list,
'cell_count': pert_cell_counts
}).sort_values('cell_count', ascending=False)
# 选一个cell丰度前25%里的扰动
top_25_percent = int(len(pert_cell_df) * 0.25)
selected_pert = pert_cell_df.iloc[top_25_percent]['perturbation']
selected_cell_count = pert_cell_df.iloc[top_25_percent]['cell_count']
print(f" - Selected perturbation: {selected_pert}")
print(f" - Cell count: {selected_cell_count}")
try:
pert_data.plot_umap(
pert=selected_pert,
point_size=20,
color=['perturbation_group'],
return_adata=False
)
except Exception as e:
print(f" - UMAP generation failed: {e}")
else:
print(" - No perturbation data available for UMAP plot.")
# ======= 4. 不画图,只保留详细统计信息 =======
# 汇报数据信息
print("\n" + "=" * 60)
print("DETAILED PERTURBATION ANALYSIS")
print("=" * 60)
# Top 10 perturbations by cell count
print("\nTop 10 perturbations by cell count:")
pert_cell_df = pd.DataFrame({
'perturbation': pert_data.filter_perturbation_list,
'cell_count': pert_cell_counts
}).sort_values('cell_count', ascending=False)
top_10_perts = pert_cell_df.head(10)
for idx, row in top_10_perts.iterrows():
print(f" {row['perturbation']}: {row['cell_count']} cells")
# Perturbation type distribution
print(f"\nPerturbation type distribution:")
pert_type_counts = pert_data.adata_split.obs['perturbation_new'].value_counts()
for pert_type, count in pert_type_counts.items():
print(f" {pert_type}: {count} cells")
# Cell type distribution
print(f"\nCell type distribution:")
celltype_counts = pert_data.adata_split.obs['celltype_new'].value_counts()
for celltype, count in celltype_counts.items():
print(f" {celltype}: {count} cells")
print("\nAnalysis completed!")
1. Analyzing cell count distribution...
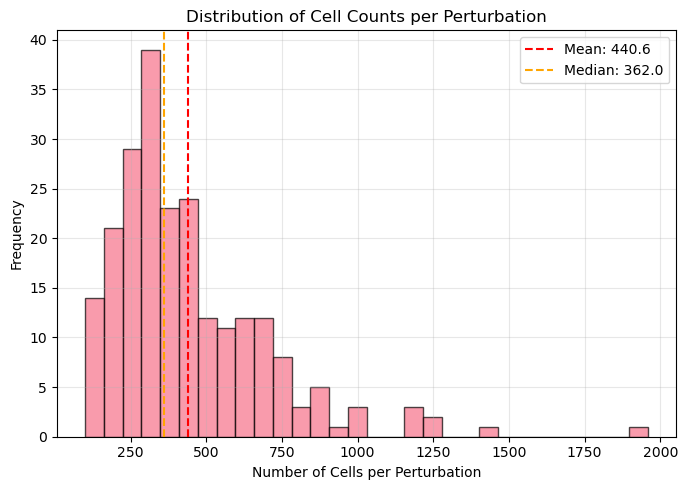
- Mean cells per perturbation: 440.6
- Median cells per perturbation: 362.0
- Total perturbations: 224
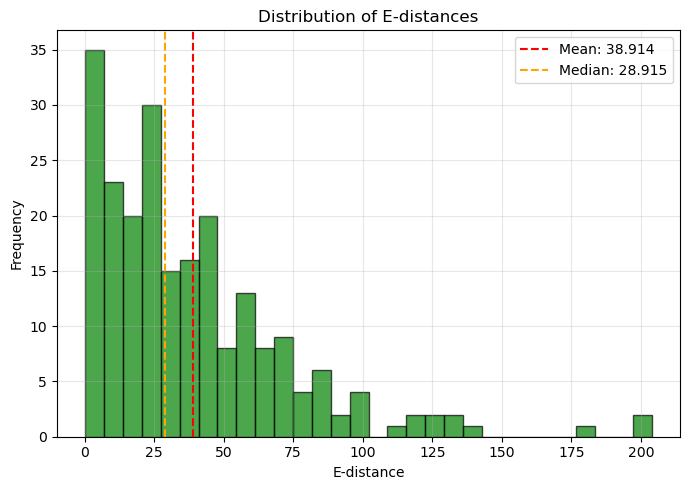
2. Analyzing E-distance distribution...
- Mean E-distance: 38.914
- Median E-distance: 28.915
- Min E-distance: 0.285
- Max E-distance: 203.848
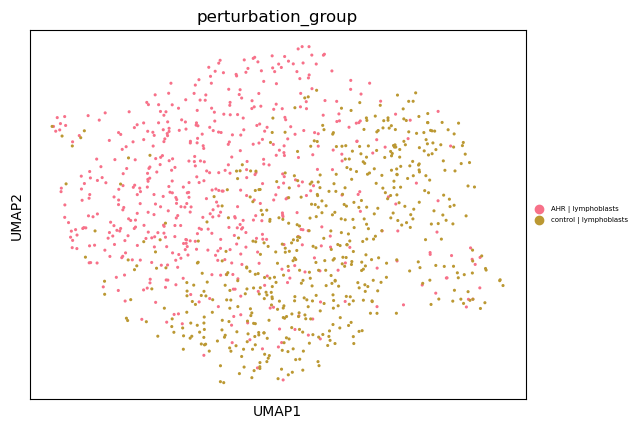
3. Generating UMAP plot for specific perturbation...
- Selected perturbation: AHR | lymphoblasts
- Cell count: 558
/nfs/public/lichen/software/miniconda3/envs/scGPT_2/lib/python3.10/site-packages/anndata/_core/anndata.py:1818: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
/nfs/public/lichen/software/miniconda3/envs/scGPT_2/lib/python3.10/site-packages/anndata/_core/anndata.py:1818: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
/nfs/public/lichen/software/miniconda3/envs/scGPT_2/lib/python3.10/site-packages/scanpy/preprocessing/_simple.py:842: UserWarning: Received a view of an AnnData. Making a copy.
view_to_actual(adata)
/nfs/public/lichen/software/miniconda3/envs/scGPT_2/lib/python3.10/site-packages/anndata/_core/anndata.py:1818: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
/nfs/public/lichen/software/miniconda3/envs/scGPT_2/lib/python3.10/site-packages/scanpy/plotting/_tools/scatterplots.py:391: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
============================================================
DETAILED PERTURBATION ANALYSIS
============================================================
Top 10 perturbations by cell count:
KLF1 | lymphoblasts: 1960 cells
BAK1 | lymphoblasts: 1457 cells
CEBPE | lymphoblasts: 1233 cells
CEBPE_RUNX1T1 | lymphoblasts: 1219 cells
UBASH3B | lymphoblasts: 1202 cells
ETS2 | lymphoblasts: 1201 cells
TBX3_TBX2 | lymphoblasts: 1167 cells
OSR2 | lymphoblasts: 1003 cells
SLC4A1 | lymphoblasts: 1000 cells
SET | lymphoblasts: 986 cells
Perturbation type distribution:
control: 11855 cells
KLF1: 1960 cells
BAK1: 1457 cells
CEBPE: 1233 cells
CEBPE_RUNX1T1: 1219 cells
UBASH3B: 1202 cells
ETS2: 1201 cells
TBX3_TBX2: 1167 cells
OSR2: 1003 cells
SLC4A1: 1000 cells
SET: 986 cells
ELMSAN1: 937 cells
ETS2_CNN1: 905 cells
MAP2K6: 878 cells
FOXF1: 874 cells
C19orf26: 872 cells
FOXA1: 851 cells
UBASH3A: 819 cells
UBASH3B_OSR2: 796 cells
DUSP9_ETS2: 787 cells
RUNX1T1: 779 cells
MEIS1: 776 cells
MAPK1: 765 cells
CNN1: 765 cells
MAP7D1: 751 cells
ZBTB25: 740 cells
DLX2: 732 cells
DUSP9: 731 cells
FEV: 704 cells
HOXB9: 701 cells
ZBTB1: 693 cells
TBX3: 691 cells
TBX2: 690 cells
PRTG: 685 cells
IKZF3: 681 cells
CLDN6: 670 cells
SET_KLF1: 667 cells
CBL: 663 cells
CBFA2T3: 658 cells
SET_CEBPE: 658 cells
FOXL2: 652 cells
ZNF318: 650 cells
LHX1_ELMSAN1: 647 cells
TGFBR2: 646 cells
KIF2C: 634 cells
FOSB: 630 cells
MAML2: 626 cells
LYL1_IER5L: 623 cells
ZC3HAV1_HOXC13: 618 cells
SPI1: 613 cells
IGDCC3: 613 cells
COL2A1: 598 cells
BCL2L11: 582 cells
ISL2: 582 cells
MAP2K3_ELMSAN1: 581 cells
MAP2K3: 579 cells
MAP2K3_MAP2K6: 571 cells
SLC6A9: 558 cells
AHR: 558 cells
UBASH3B_UBASH3A: 554 cells
CDKN1B: 550 cells
TMSB4X: 543 cells
S1PR2: 541 cells
BCORL1: 533 cells
ZC3HAV1: 516 cells
MAPK1_PRTG: 500 cells
SAMD1: 498 cells
MAPK1_TGFBR2: 497 cells
ARRDC3: 495 cells
FOXA3: 494 cells
CEBPB: 488 cells
SGK1: 483 cells
AHR_KLF1: 481 cells
CSRNP1: 478 cells
CELF2: 475 cells
CNNM4: 470 cells
CEBPE_KLF1: 468 cells
SNAI1: 467 cells
BPGM: 464 cells
GLB1L2: 463 cells
BCL2L11_TGFBR2: 463 cells
CEBPA: 460 cells
ETS2_MAPK1: 459 cells
MAP2K6_ELMSAN1: 451 cells
ETS2_IKZF3: 445 cells
PTPN12: 445 cells
ETS2_PRTG: 445 cells
PTPN9: 445 cells
IRF1_SET: 444 cells
ETS2_IGDCC3: 441 cells
TSC22D1: 441 cells
LYL1: 432 cells
IRF1: 427 cells
FOSB_UBASH3B: 426 cells
PTPN1: 419 cells
CEBPE_PTPN12: 415 cells
CEBPB_MAPK1: 412 cells
CBL_UBASH3B: 410 cells
ZC3HAV1_CEBPE: 410 cells
IER5L: 405 cells
UBASH3B_CNN1: 402 cells
CNN1_MAPK1: 398 cells
KLF1_BAK1: 395 cells
HOXC13: 385 cells
FOSB_OSR2: 382 cells
DUSP9_IGDCC3: 382 cells
ATL1: 379 cells
LHX1: 375 cells
IGDCC3_MAPK1: 368 cells
DUSP9_KLF1: 366 cells
MAP4K5: 366 cells
PTPN12_PTPN9: 364 cells
MAP4K3: 363 cells
KLF1_FOXA1: 361 cells
CDKN1A: 360 cells
POU3F2: 359 cells
BPGM_ZBTB1: 358 cells
SGK1_S1PR2: 358 cells
ETS2_CEBPE: 354 cells
SAMD1_PTPN12: 351 cells
SGK1_TBX2: 349 cells
CBL_CNN1: 348 cells
FOSB_PTPN12: 345 cells
PTPN12_OSR2: 339 cells
MAP2K3_SLC38A2: 338 cells
KLF1_TGFBR2: 337 cells
CBL_PTPN12: 335 cells
SLC38A2: 332 cells
CEBPB_PTPN12: 331 cells
RREB1: 331 cells
FOXA3_HOXB9: 329 cells
TMSB4X_BAK1: 329 cells
COL1A1: 328 cells
RHOXF2: 328 cells
TGFBR2_C19orf26: 325 cells
UBASH3B_PTPN9: 325 cells
ETS2_MAP7D1: 324 cells
EGR1: 322 cells
RHOXF2_SET: 321 cells
TGFBR2_ETS2: 318 cells
PTPN12_UBASH3A: 311 cells
KLF1_CEBPA: 311 cells
STIL: 309 cells
POU3F2_FOXL2: 309 cells
CBL_PTPN9: 306 cells
PTPN12_ZBTB25: 303 cells
HK2: 303 cells
MAPK1_IKZF3: 302 cells
MAP2K6_SPI1: 302 cells
BPGM_SAMD1: 301 cells
TGFBR2_IGDCC3: 301 cells
MAP2K6_IKZF3: 300 cells
SGK1_TBX3: 300 cells
SNAI1_UBASH3B: 299 cells
KLF1_COL2A1: 298 cells
UBASH3B_PTPN12: 298 cells
MIDN: 298 cells
PRDM1: 297 cells
DUSP9_PRTG: 297 cells
FOXA1_HOXB9: 295 cells
DUSP9_MAPK1: 290 cells
FEV_ISL2: 284 cells
PTPN12_SNAI1: 283 cells
KIAA1804: 278 cells
FEV_MAP7D1: 277 cells
AHR_FEV: 276 cells
HOXA13: 274 cells
FOXA1_FOXF1: 272 cells
PTPN13: 270 cells
JUN: 266 cells
ZBTB10_PTPN12: 265 cells
TGFBR2_PRTG: 265 cells
C3orf72: 264 cells
KLF1_MAP2K6: 263 cells
RHOXF2_ZBTB25: 263 cells
MAP2K3_IKZF3: 261 cells
UBASH3B_ZBTB25: 255 cells
KIF18B: 254 cells
TP73: 248 cells
FOSB_IKZF3: 247 cells
ZNF318_FOXL2: 246 cells
CKS1B: 241 cells
KMT2A: 241 cells
SAMD1_UBASH3B: 240 cells
NCL: 237 cells
CEBPE_CNN1: 237 cells
HNF4A: 237 cells
KLF1_CLDN6: 237 cells
ARID1A: 232 cells
CDKN1C: 230 cells
FOXA3_FOXA1: 216 cells
POU3F2_CBFA2T3: 216 cells
CITED1: 215 cells
FOXO4: 215 cells
CEBPB_OSR2: 211 cells
FOXF1_HOXB9: 200 cells
FOXL2_MEIS1: 193 cells
NIT1: 192 cells
LYL1_CEBPB: 192 cells
FOXA1_FOXL2: 192 cells
FOXF1_FOXL2: 192 cells
SAMD1_ZBTB1: 191 cells
CBL_TGFBR2: 188 cells
DUSP9_SNAI1: 183 cells
ZBTB10_ELMSAN1: 182 cells
CEBPE_CEBPA: 180 cells
CEBPE_SPI1: 179 cells
CNN1_UBASH3A: 178 cells
BCL2L11_BAK1: 175 cells
FOXA3_FOXF1: 175 cells
ZBTB10: 162 cells
FEV_CBFA2T3: 159 cells
FOSB_CEBPE: 151 cells
IGDCC3_ZBTB25: 141 cells
IGDCC3_PRTG: 140 cells
CDKN1B_CDKN1A: 134 cells
SNAI1_DLX2: 131 cells
FOXA3_FOXL2: 130 cells
HES7: 126 cells
CDKN1C_CDKN1A: 126 cells
CEBPE_CEBPB: 114 cells
PLK4: 113 cells
CDKN1C_CDKN1B: 107 cells
PRDM1_CBFA2T3: 106 cells
ZBTB10_SNAI1: 100 cells
Cell type distribution:
lymphoblasts: 110554 cells
Analysis completed!
[ ]:
[ ]: